3. Protein Design
Part A - Protein Analysis
How many molecules of amino acids do you take with a piece of 500 grams of meat? (on average an amino acid is ~100 Daltons)
Different “meats” have different concentrations of proteins-fat. So according to USDA, “Pork, fresh, loin, tenderloin, separable lean only, cooked, roasted” has 26.17% Briefly describe the protein you selected and why you selected it. protein. So 500 grams have 130.85grams of proteins. Each amino acid is ~100 Daltons which is 1.66e-22grams. So the number of amino acids is 7.882e23 - which is 1.31mole (divide by Avogadro’s number - 6.022e23)
Why are there only 20 natural amino acids?
Hard question… theoretically there could by 64 (4^3) amino acids. Nature leans to robustness, so having a few different codons coding for the same amino acids gives some variance and robustness to mutations. Not sure about the macgic number 20 (and there a few organisms that use more than 20, but very rarely?)
Why most molecular helices are right handed?
Made up of L-amino acids, the right handed conformations produces less steric clashes and hindrances.

Where did amino acids come from before enzymes that make them, and before life started?
From Wikipedia:
The Miller–Urey experiment[1] (or Miller experiment)[2] was a chemical experiment that simulated the conditions thought at the time (1952) to be present on the early Earth and tested the chemical origin of life under those conditions. The experiment at the time supported Alexander Oparin's and J. B. S. Haldane's hypothesis that putative conditions on the primitive Earth favoured chemical reactions that synthesized more complex organic compounds from simpler inorganic precursors. Considered to be the classic experiment investigating abiogenesis, it was conducted in 1952[3] by Stanley Miller, with assistance from Harold Urey, at the University of Chicago and later the University of California, San Diego and published the following year.[4][5][6]
After Miller's death in 2007, scientists examining sealed vials preserved from the original experiments were able to show that there were actually well over 20 different amino acids produced in Miller's original experiments. That is considerably more than what Miller originally reported, and more than the 20 that naturally occur in life.[7] More recent evidence suggests that Earth's original atmosphere might have had a composition different from the gas used in the Miller experiment, but prebiotic experiments continue to produce racemic mixtures of simple-to-complex compounds under varying conditions
What do digital databases and nucleosomes have in common?
Both compress a large amount of information, to fit in a small space but still able to be retrievable when needed.
Pick any protein (from any organism) of your interest that has a 3D structure and answer the following questions.
Briefly describe the protein you selected and why you selected it.
I was curious to see which structures were already figured out for COVID-19 (2019-nCov - “Coronavirus”). A short search in RCSB and I found:
6LU7 - The crystal structure of COVID-19 main protease in complex with an inhibitor N3
Identity the amino acid sequence of your protein.
- How long is it? What is the most frequent amino acid?
306aa
{'S': 16,
'G': 26,
'F': 17,
'R': 11,
'K': 11,
'M': 10,
'A': 17,
'P': 13,
'V': 27,
'E': 9,
'C': 12,
'Q': 14,
'T': 24,
'L': 29,
'N': 21,
'W': 3,
'D': 17,
'Y': 11,
'H': 7,
'I': 11}
The most common aa is Leucine - 29/306
- How many protein sequence homologs are there for your protein?
There are 98 sequences in the blastp return results, but around 30~ are exactly the same.
- Does your protein belong to any protein family?
Identify the structure page of your protein in RCSB
http://www.rcsb.org/structure/6LU7
-
When was the structure solved? 2020-01-26
-
Is it a good quality structure? Resolution: 2.16 Å
-
Are there any other molecules in the solved structure apart from protein?
Yes: N-[(5-METHYLISOXAZOL-3-YL)CARBONYL]ALANYL-L-VALYL-N~1~-((1R,2Z)-4-(BENZYLOXY)-4-OXO-1-{[(3R)-2-OXOPYRROLIDIN-3-YL]METHYL}BUT-2-ENYL)-L-LEUCINAMIDE
- Does your protein belong to any structure classification family?
Open the structure of your protein in any 3D molecule visualization software
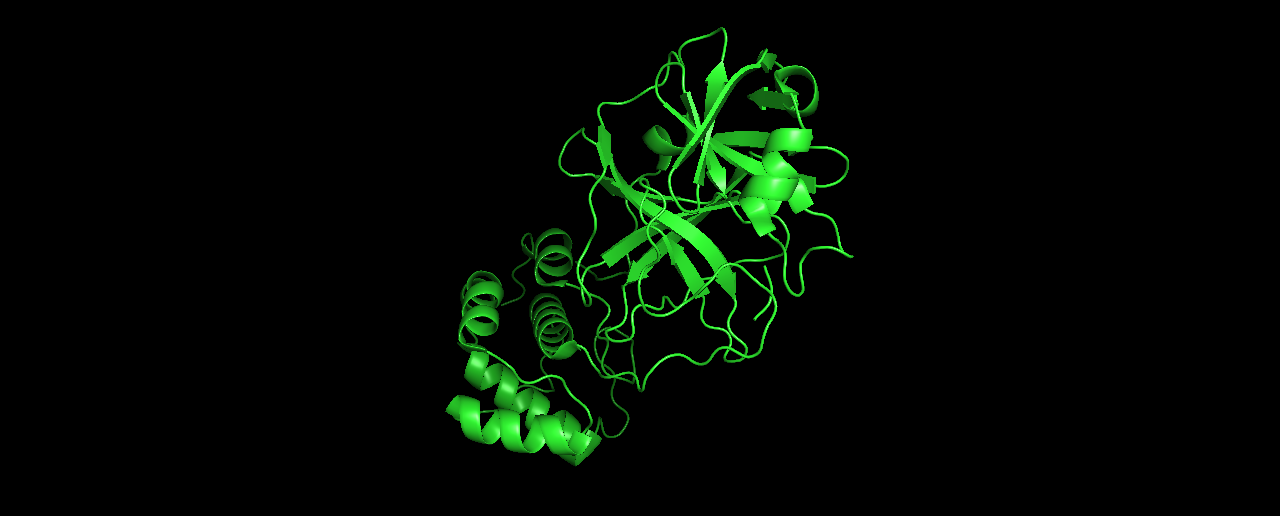
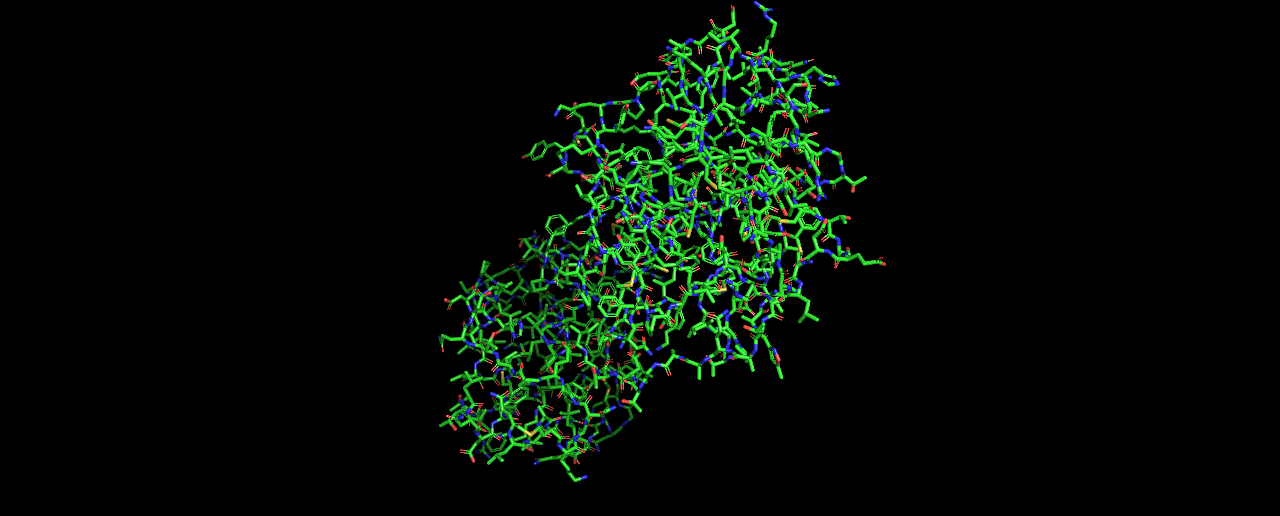
- Visualize the protein as “cartoon”, “ribbon” and “ball and stick”.
Cartoon:
Ribbon:

Ball and stick:
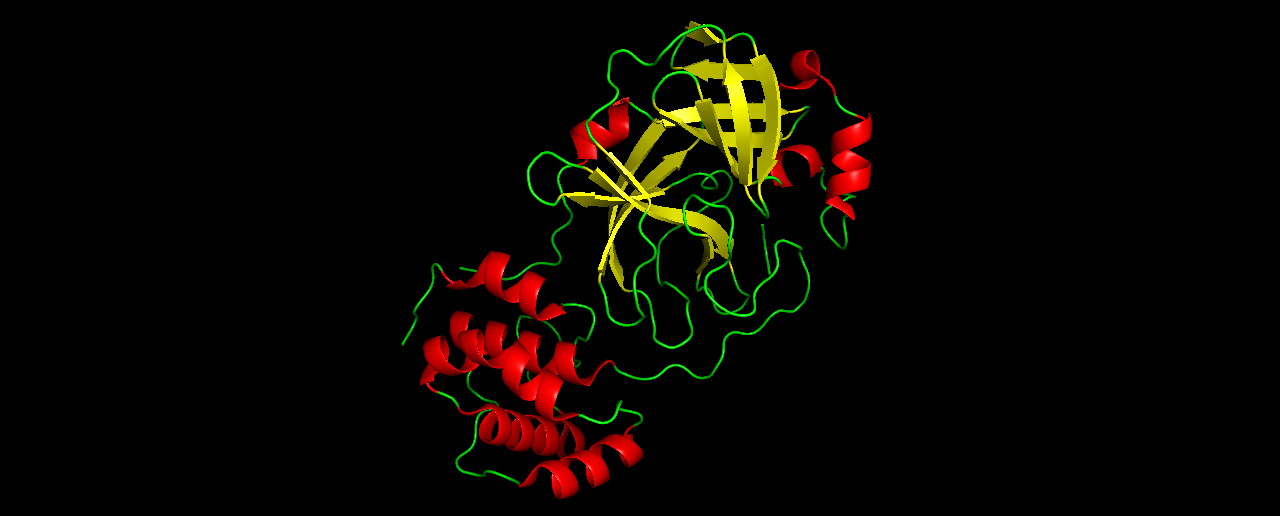
- Color the protein by secondary structure. Does it have more helices or sheets?
Similar amount of helices and sheets
- Color the protein by residue type. What can you tell about the distribution of hydrophobic vs hydrophilic residues?
To do this, I followed the instructions here, and got:
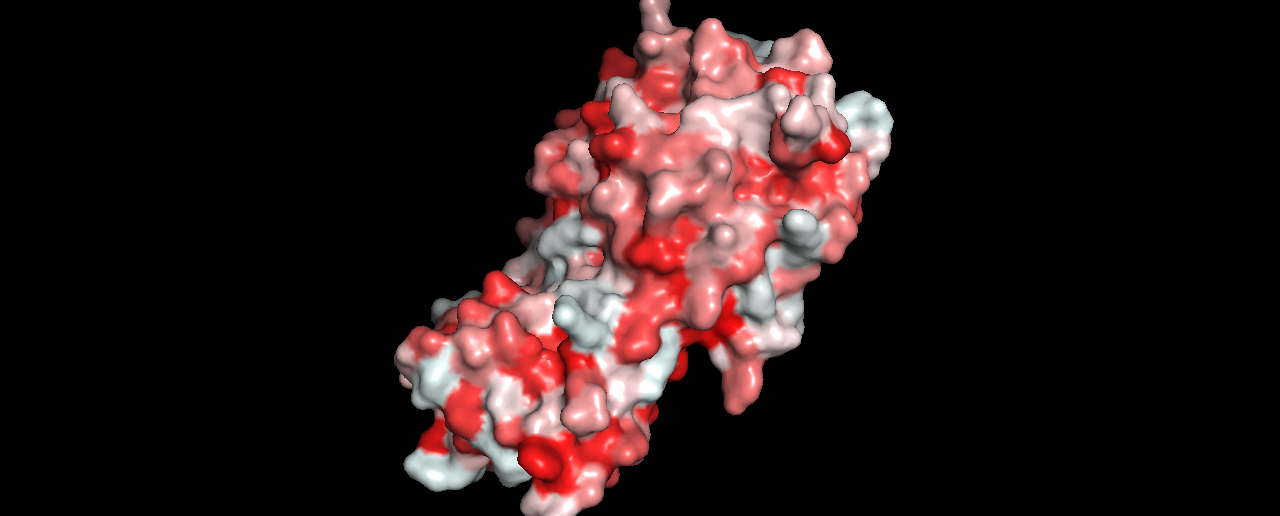
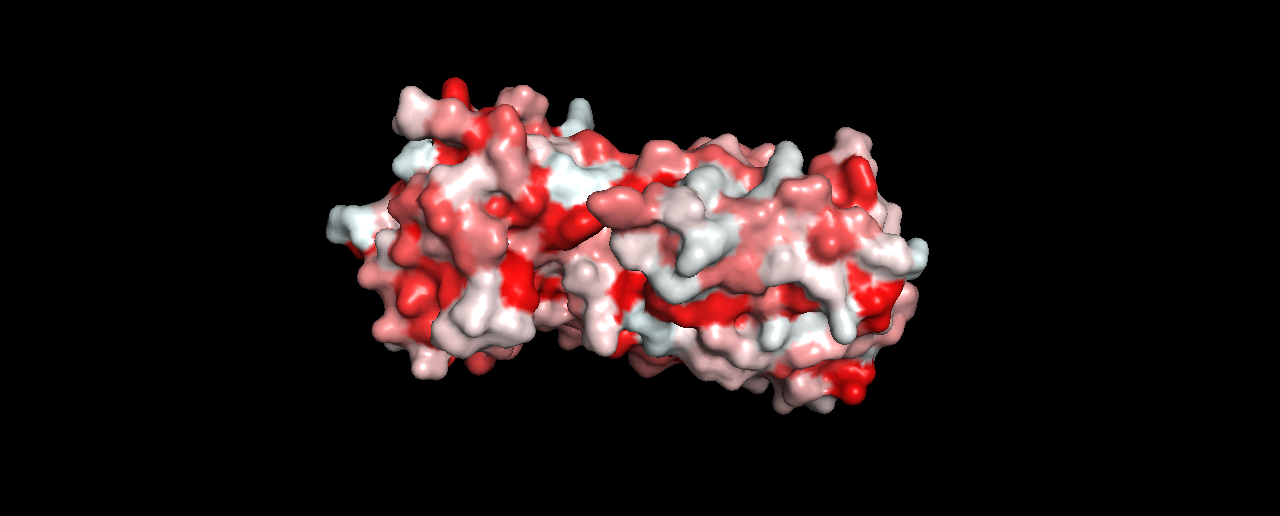
- Visualize the surface of the protein. Does it have any “holes” (aka binding pockets)?
Not sure if this is enough to be considered a binding pocket?
Part B - How to (almost) Fold (almost) Anything
Folding a small (30 aa) peptide.
Click here to open the notebook
Pick the lowest energy model and structurally (visually) compare it to the native. How close is it to the native? If its different, what parts did the computer program get wrong?
Alignment results (true structure in Cyan):
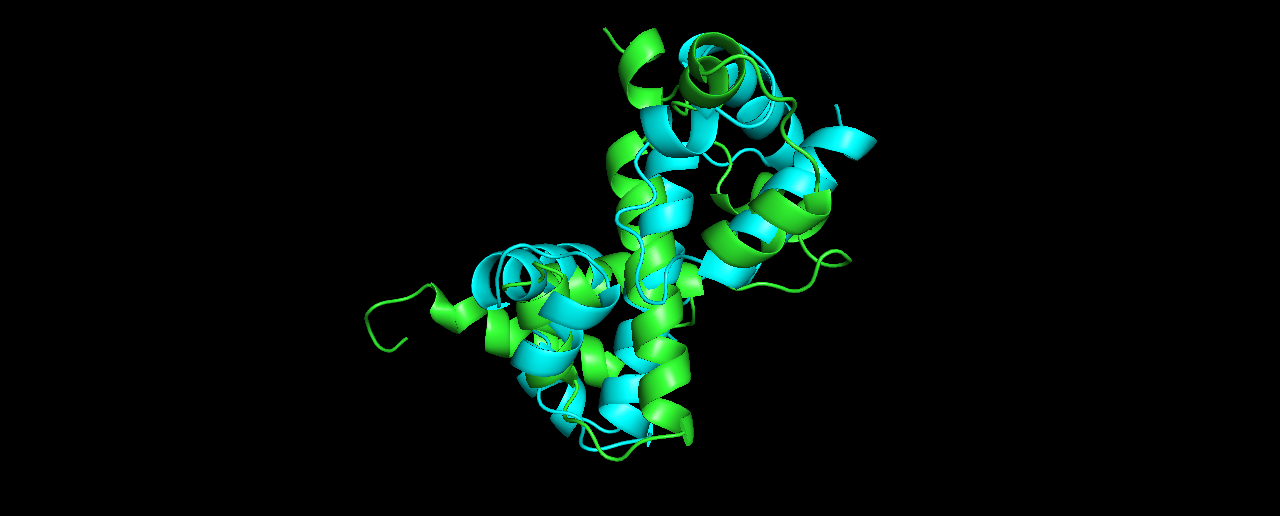
The alignment is not so good… at which parts?
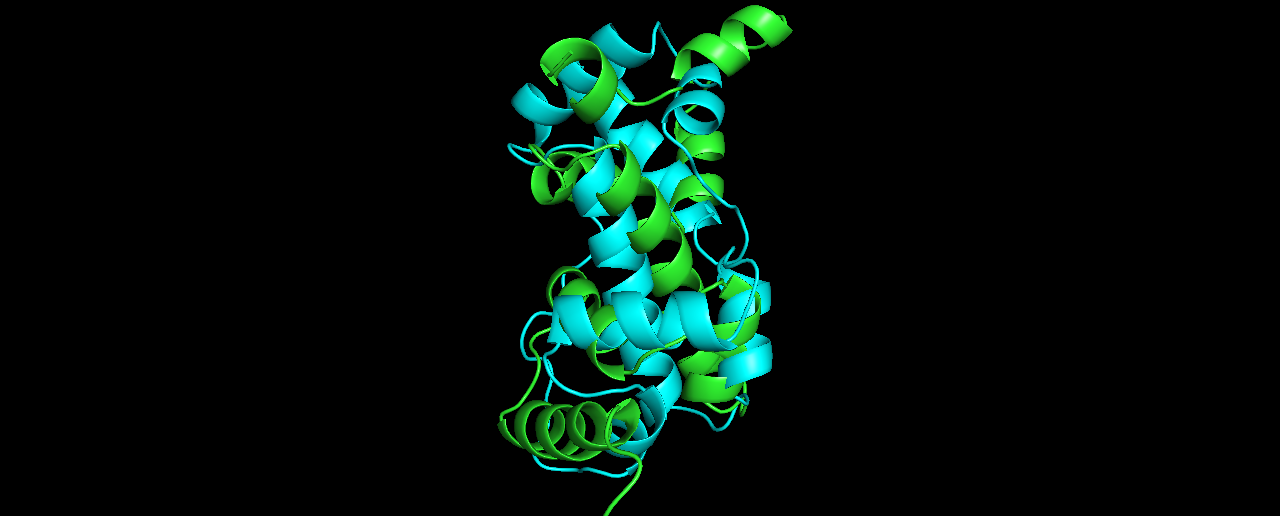
Pick the lowest RMSD model and structurally compare it to the native. How close is it to the native? If its different than the lowest energy model, how is it different?
Alignment results (true structure in Cyan):
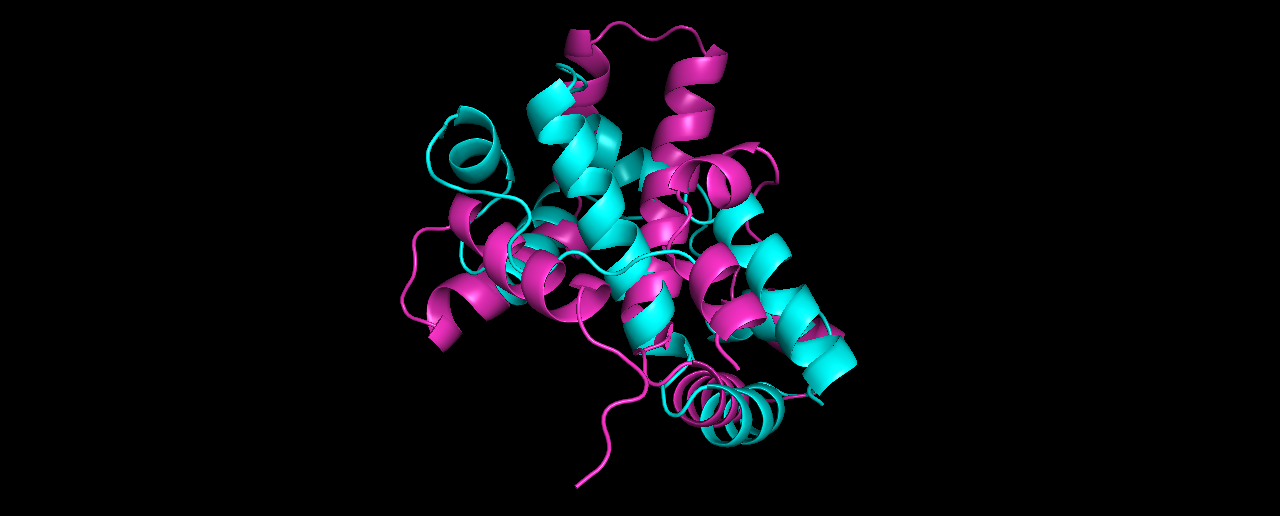
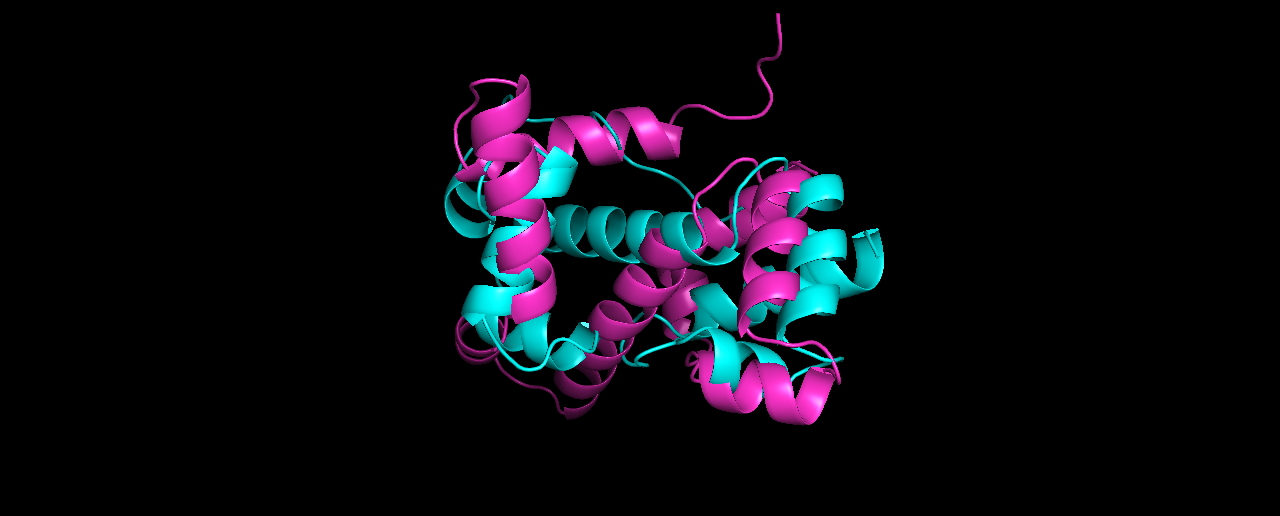
The lowest RMSD model (8.27) is quite different from the lowest energy one, although I expected it to be more similar to the true solved structure than it looks like.
Folding a random peptide
Click here to open the notebook
To summarize, I followed the same steps in the Notebook, but instead of this line:
pose = prs.pose_from_sequence(native_pose.sequence())
with
pose = prs.pose_from_sequence(sequence)
To generate a random sequence, I wrote a simple generator that generates an N-long string, given a given probability of letters:
# generate a random aa sequence with the same aa distribution
N = 50
# taken from https://web.expasy.org/protscale/pscale/A.A.Swiss-Prot.html
aa_freqs = {
'A': 8.25,
'R': 5.53,
'N': 4.06,
'D': 5.45,
'C': 1.37,
'Q': 3.93,
'E': 6.75,
'G': 7.07,
'H': 2.27,
'I': 5.96,
'L': 9.66,
'K': 5.84,
'M': 2.42,
'F': 3.86,
'P': 4.70,
'S': 6.56,
'T': 5.34,
'W': 1.08,
'Y': 2.92,
'V': 6.87
}
s = ""
for aa, freq in aa_freqs.items():
for i in range(int(freq * 20)):
s += aa
sequence = ""
indices = np.random.randint(0, len(s) - 1, N)
for idx in indices:
sequence += s[idx]
Then, we also need to choose a new fragment library. First, I used the data/random library that was supplied to us. Here are is the list of folds with lowest energies:
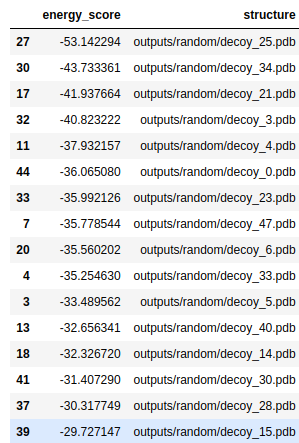
And here are the top-3 structures:


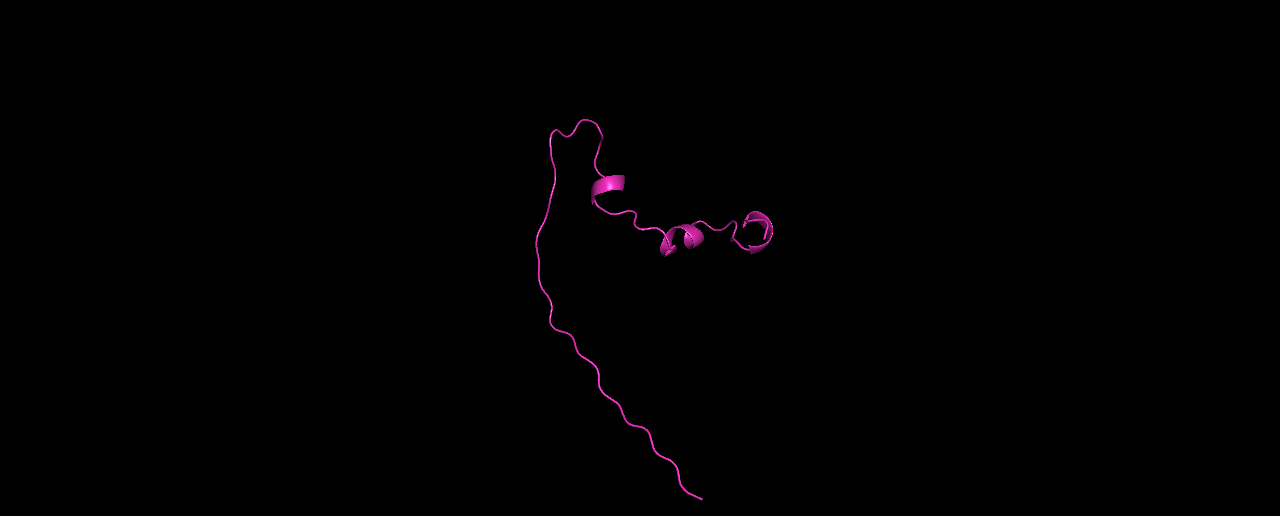
In my opinion, these structures don’t look very protein-like. The linear tail and the low percentage of sheets/helices is unusual.
Second try, I now ran the exact same sequence with a fragment library generated by Robetta. The folds gave a lower energy score compared to the one above:
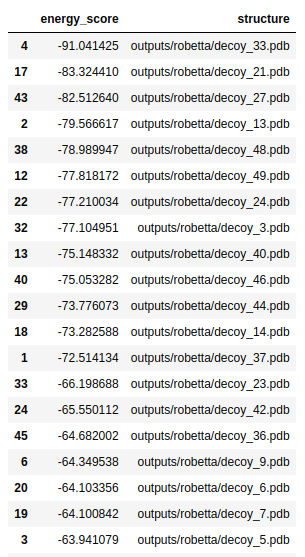
And the top-3:
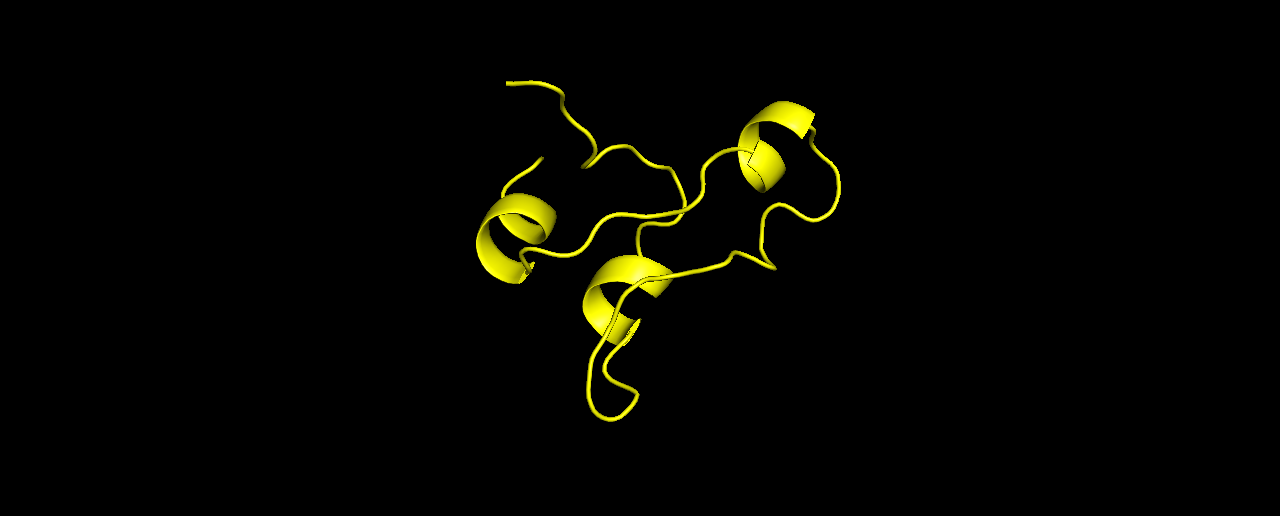
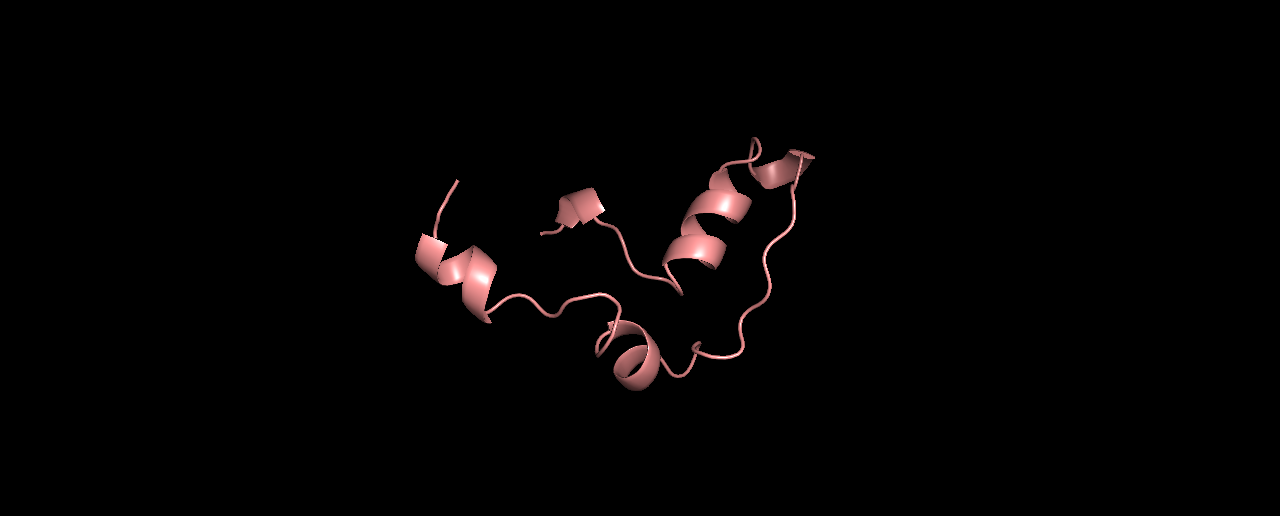
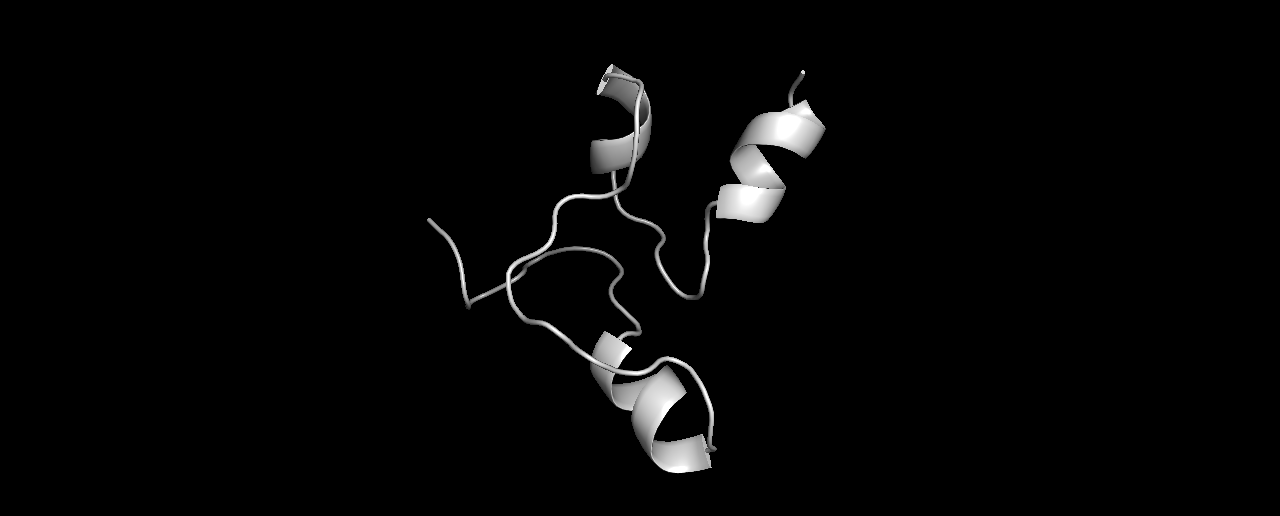
Looking much better! it seems the odd linear tail was caused by using the wrong library. And the folds make more “sense” now.
How to Fold Your Name
Amino acids are “just” letters, so what can we write using this alphabet? Practicing casual narcissism, I wanted to created a protein with only my name. As we saw, creating a fragment library is essential, but Robetta decided that my name is not fragmentable:

The error is sam target99 failed! - which means that the secondary structure prediction failed. Sad.
Also the sequence EYALEYAALEYAAALEYAAAALEYAAAAAL failed.
Alas, the sequence EYALPERRYEYALPERRYEYALPERRYEYALPERRY worked! Here are the top 3 structures:
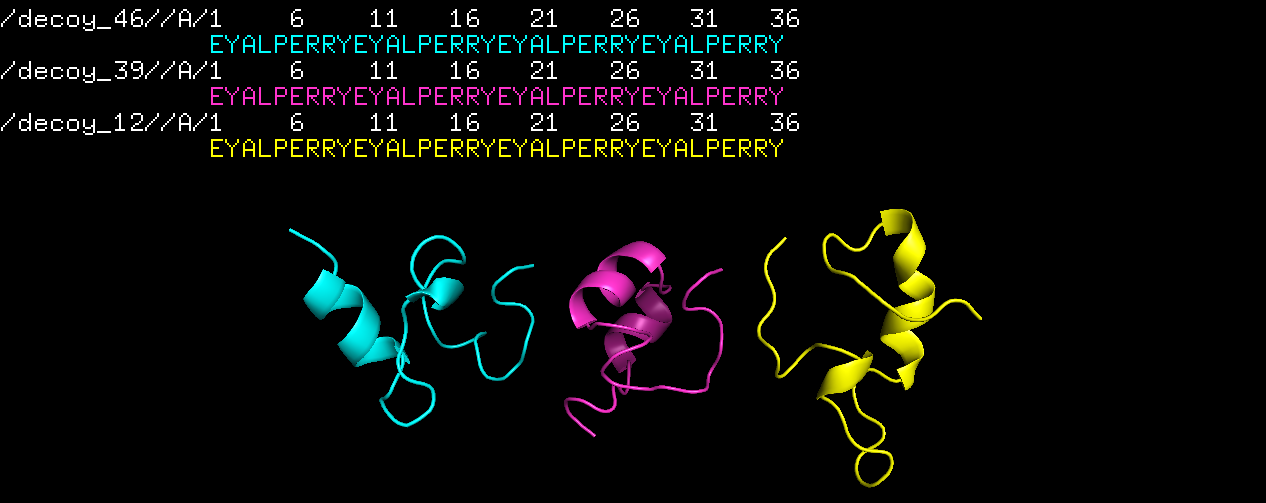
Exciting.
Even more exciting (and harmonious) is a protein structure made by whole of the names of the HTGAA cohort. See it at Belen’s page!